Medical device software now powers virtually every category of medical device. These range from Class I wearables to Class III implantable systems.
The global medical device market is projected to reach USD 1032.66 billion by 2034, and a large share of the industry's growth is driven by software. Right from AI diagnostic platforms to embedded device firmware, cloud-connected monitoring, and mobile clinical tools, all need access to software technology.
However, medical device software development is not a faster version of enterprise healthcare software development. It carries patient safety liability, regulatory filing requirements, and post-market surveillance obligations that standard engineering practices don't address.
Also, most delays trace back to documentation gaps. These are incomplete integrations of risk management or architectural decisions made without regulatory input.
This guide covers the full development process along with the compliance standards that govern it. Furthermore, it identifies the obstacles that can derail regulatory submissions. It will also define important architectural and resource allocation decisions for success.
What is Medical Device Software Development?
Medical device software development involves designing, implementing, verifying, and maintaining software. The software is either embedded within a physical medical device (SiMD) or operates independently on general hardware. These are designed to serve a medical purpose (SaMD), subject to IEC 62304 and applicable FDA or EU MDR requirements.
Types of Medical Device Software
Discover the four categories of medical device software and how they enhance clinical and patient care processes.
1. Embedded Software (SiMD)
These are firmware that directly controls hardware. Device examples include pacemakers, infusion pump controllers, and ventilators. The primary standards are IEC 62304 and IEC 60601-1.
2. Software as a Medical Device (SaMD)
SaMD is standalone software on general hardware that serves a medical purpose. Some of the quick examples include AI imaging analysis platforms and clinical decision support tools. The primary standards for development are FDA SaMD Guidance and IEC 62304.
3. Connected Device Platforms
These are IoT-integrated devices that transmit clinical data to cloud infrastructure. The primary standards followed are IEC 62304 and IEC 62443 (cybersecurity).
4. Clinical Decision Support (CDS)
CDS are algorithms that recommend treatment, flag risk, or interpret diagnostic data. The primary standard involves FDA CDS Guidance (2022, updated 2025).
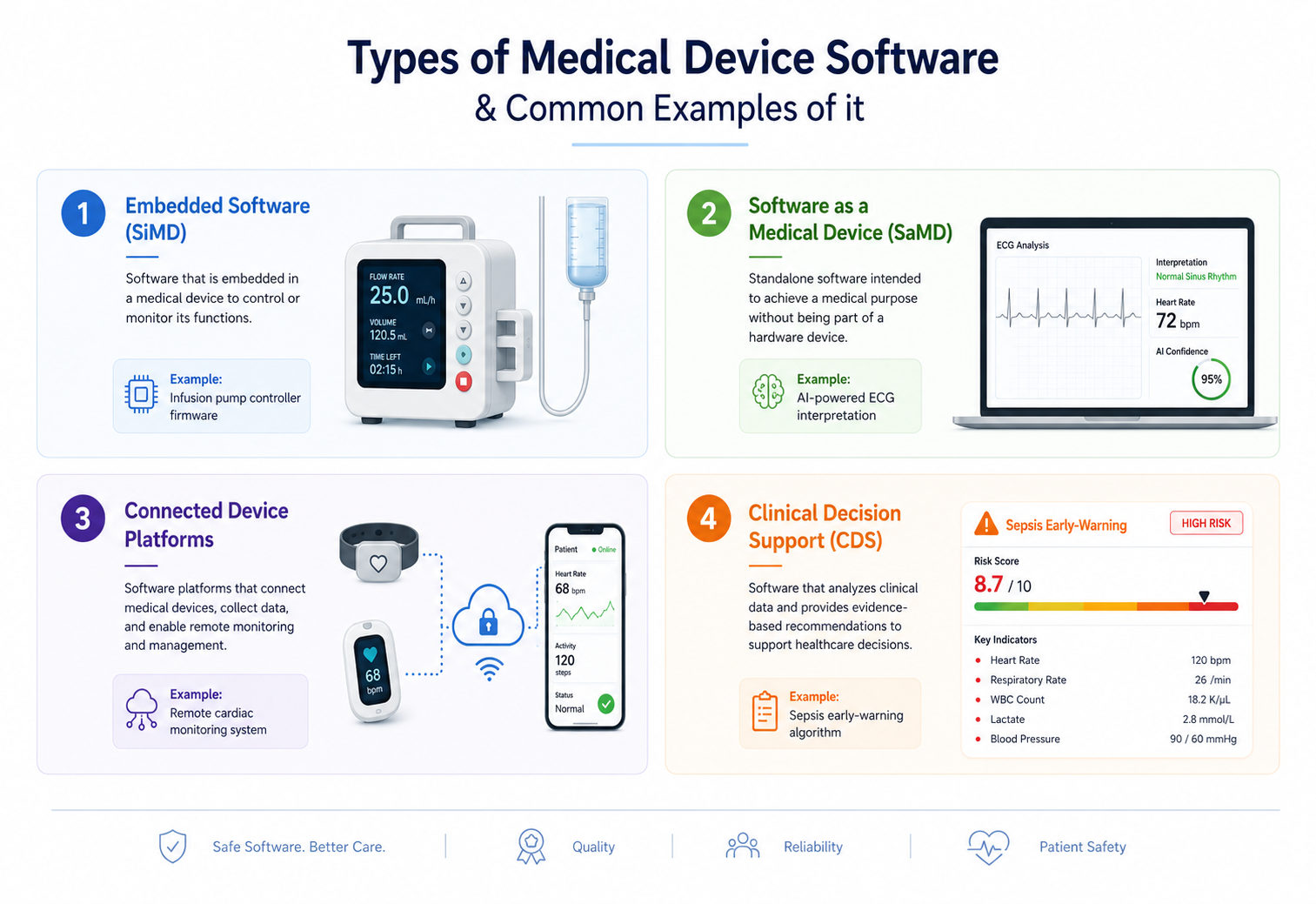
Common Examples of Medical Device Software
|
Software Type |
Example Application |
Regulatory Impact |
|
Embedded (SiMD) |
Infusion pump controller firmware |
IEC 62304 Class C, full SDLC documentation |
|
SaMD |
AI-powered ECG interpretation |
FDA SaMD pathway, PCCP if adaptive |
|
Connected Platform |
Remote cardiac monitoring system |
IEC 62443 SBOM, threat modeling required |
|
Clinical Decision Support |
Sepsis early-warning algorithm |
FDA CDS Guidance, intended use classification |
Why Medical Device Software Development is different from Traditional Software?
Traditional software ships and iterates. Medical device software ships after documented verification and validation, and every post-market change requires a defined change control process. A defect's cost is measured in patient outcomes, not sprint velocity.
a) Patient safety liability
Software defects can cause direct harm. IEC 62304 and ISO 14971 make risk identification a mandatory engineering activity, not a retrospective one.
b) Regulatory compliance drives architecture
Software safety classification under IEC 62304 determines the depth of documentation, testing rigor, and change control, the scope before the first line of code is written.
c) Real-time and reliability requirements
Embedded device software often runs on RTOS platforms where latency tolerance is measured in milliseconds. A delayed interrupt in a ventilator controller is not a UX problem.
d) Cybersecurity is now a submission requirement
Following the FDA's Cybersecurity Guidance, enforced on all submissions since October 2023, a Software Bill of Materials (SBOM) and documented threat model are mandatory. Submissions missing either are returned before substantive review begins.
e) AI and ML validation have no deterministic baseline
Probabilistic outputs require performance thresholds by patient subgroup, documentation of training data provenance, and a Predetermined Change Control Plan (PCCP) under the FDA's 2025 guidance for any adaptive component.
The Medical Device Software Development Process
Medical device software follows a V-Model lifecycle where every development phase has a corresponding verification or validation counterpart. Skipping phases generates regulatory debt that compounds at submission.
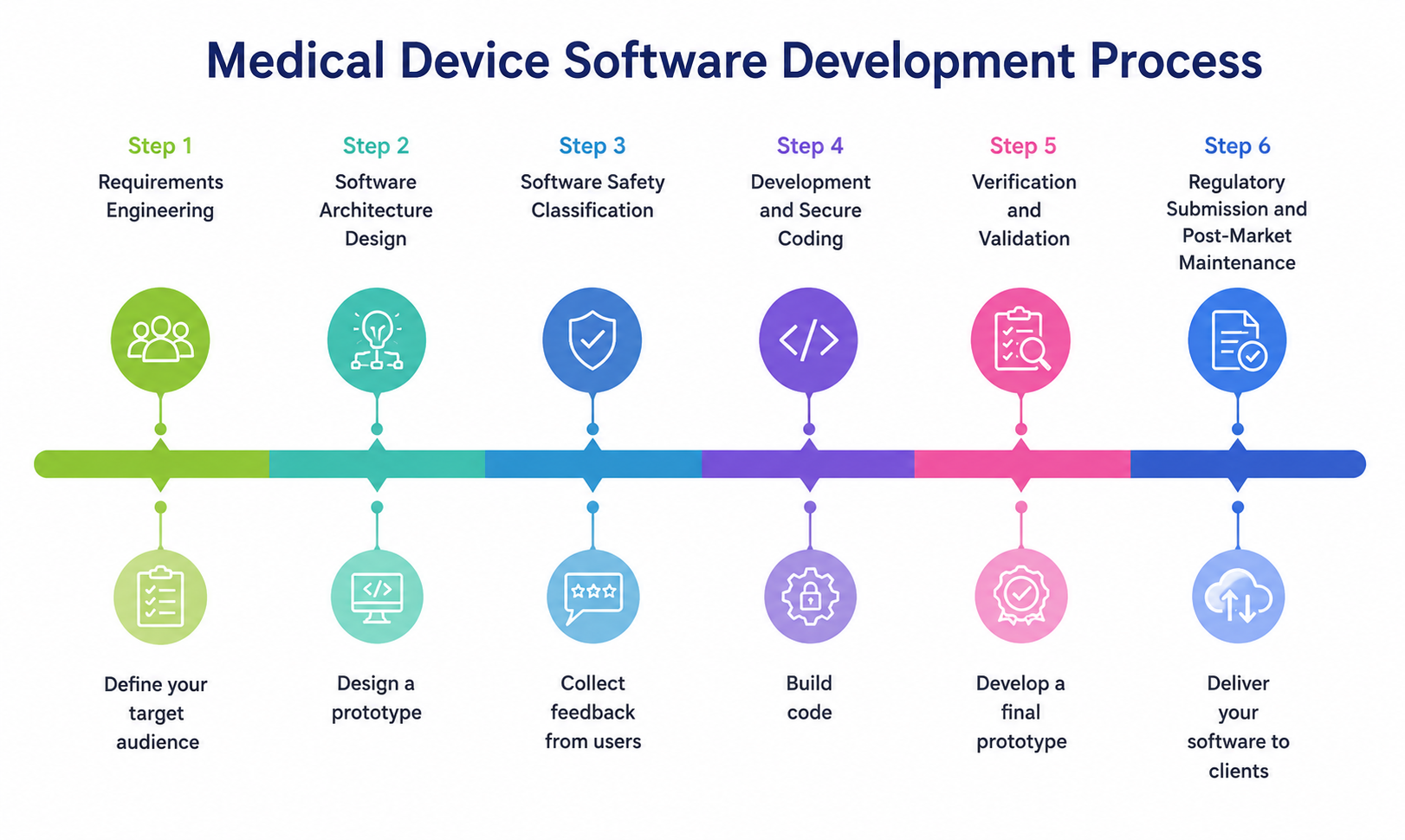
Step 1: Requirements Engineering
Requirements are the most consequential phase and the most commonly under-resourced. Poorly defined requirements cause architecture changes after design sign-off, which is the most expensive category of engineering rework.
-
Clinical Requirements Gathering: Clinical requirements define intended use, patient population, and contraindications. These feed directly into software safety classification and risk analysis. Gaps here produce misclassified software and incomplete hazard identification downstream.
-
Regulatory Requirements Mapping: Map intended use against applicable standards before architecture begins. These are IEC 62304, ISO 14971, FDA guidance, and EU MDR Annex I. These are mandatory. Any team that tries to retrofit compliance requirements after development routinely adds 4 to 8 months to submission timelines.
-
User and Stakeholder Requirements: IEC 62366 usability engineering requires documented user needs and use scenarios before final design. FDA expects a usability file with any Class II or III device submission.
Step 2: Software Architecture Design
Architecture is where compliance costs get set. The right layering produces testable components with clean audit trails. The wrong layering produces untestable modules and retroactive documentation debt.
Reference Architecture: Six-Layer Model for Class B/C Devices
-
Device Layer: Hardware platform, sensors, MCU/FPGA interfaces
-
Firmware Layer: RTOS (FreeRTOS, VxWorks, Embedded Linux), hardware abstraction
-
Connectivity Layer: Bluetooth LE, Wi-Fi, cellular, HL7 FHIR, DICOM protocol stacks
-
Cloud Infrastructure: AWS GovCloud / Azure Healthcare APIs, encrypted data pipelines
-
Analytics and Reporting: AI/ML inference, clinical dashboards, audit logging
-
EHR/EMR Integration: HL7 FHIR R4, SMART on FHIR, CDS Hooks
Compliance checkpoints sit at every layer boundary. Each interface requires a documented specification.
3 architecture decisions that directly affect regulatory cost:
1. SOUP management
Every third-party library, including open source, is Software of Unknown Provenance under IEC 62304 and requires a documented risk assessment. Teams with large dependency trees routinely underestimate SOUP documentation effort by 3x.
2. Cybersecurity architecture
SBOM generation, STRIDE threat modeling, and dependency vulnerability scanning must be defined before architecture sign-off. Retrofitting security controls after implementation costs 4 to 6 times as much as building them in (NIST Secure by Design).
3. AI component isolation
ML models must be architecturally bounded with defined performance thresholds. FDA's 2025 PCCP guidance requires that any adaptive algorithm have a documented change management protocol in the original submission.
Step 3: Software Safety Classification
|
IEC 62304 Class |
Risk Level |
Documentation Depth |
Typical Examples |
|
Class A |
No injury possible |
Minimal, basic change control |
Administrative tools, scheduling UI |
|
Class B |
Non-serious injury possible |
Full SDLC documentation, unit testing |
Patient monitoring displays, alert systems |
|
Class C |
Serious injury or death is possible |
Full SDLC + detailed design + all testing levels |
Infusion pump controllers, AI diagnostic engines |
Step 4: Development and Secure Coding
Class B and C software requires static analysis (Polyspace, Coverity, Klocwork) and MISRA C compliance for embedded code. FDA reviewers look for evidence of static analysis in test documentation. Its absence in a Class C submission triggers a Major Deficiency letter. Integrate these tools in CI/CD from sprint one, not as a pre-submission check.
Step 5: Verification and Validation
|
Dimension |
Verification |
Validation |
|
Core question |
Did we build the software right? |
Did we build the right software? |
|
What it tests |
Conformance to specified requirements |
Fitness for intended use and user needs |
|
Key activities |
Unit, integration, system testing, code review |
Usability testing, clinical simulation, UAT |
|
Who performs it |
Engineering team |
Qualified users or clinical evaluators |
|
FDA document reference |
Design verification report |
Validation protocol and report |
|
Common failure mode |
Missing traceability to requirements |
Submitted as evidence of validation (it is not) |
Step 6: Regulatory Submission and Post-Market Maintenance
-
FDA Submission Preparation: FDA's 510(k) and De Novo pathways require a documented software description. It requires hazard analysis, V&V summary, and, post-October 2023, a full cybersecurity documentation package including SBOM and threat model.
-
Post-Market Monitoring: IEC 62304 clause 6.x requires a defined change management process. Teams without this when they ship have no clear pathway for emergency patch deployment when a critical defect surfaces.
-
Software Updates and Change Management: FDA's PCCP framework allows AI components to update within pre-approved bounds without a new 510(k), but only if the change control plan was documented in the original submission.
Key Compliance Standards for Medical Device Software Development
No single standard covers medical device software compliance. FDA submissions require demonstrated conformance to multiple overlapping frameworks, each governing a different aspect of the engineering process.
|
Standard |
Scope |
What Breaks Without It |
|
IEC 62304 |
Software development lifecycle: planning through maintenance |
Missing SDLC documentation is the top 510(k) deficiency |
|
ISO 14971 |
Risk management for medical devices and software |
Inadequate risk controls were cited in 63% of FDA warning letters (FDA CDRH, 2026) |
|
ISO 13485 |
Quality management system requirements |
No documented basis for any process claim |
|
IEC 62366-1 |
Usability engineering: user needs through formative and summative testing |
Class II+ submissions require a usability file; missing it delays review |
|
IEC 62443 |
Cybersecurity for industrial and connected systems |
FDA cross-references for networked device SBOM and threat modeling |
|
FDA Cybersecurity Guidance (2023) |
SBOM, threat modeling, vulnerability disclosure for networked devices |
Non-compliant submissions rejected before substantive review since Oct 2023 |
Top Challenges in Medical Device Software Development
The most expensive challenges in this domain are process failures. They show up as regulatory rejections leading to product recalls or extended review cycles.
1. Managing Complex, Multi-Framework Regulatory Requirements
A single device shipping to the US, EU, and APAC may need to satisfy FDA 21 CFR Part 820, EU MDR 2017/745, IEC 62304, IEC 62443, ISO 14971, and IEC 62366 simultaneously. These frameworks don't fully align. Teams that build their QMS around one and retrofit the others generate inconsistent documentation that reviewers flag.
2. Maintaining End-to-End Traceability
Every requirement must trace to a design element. Every design element must trace to a test. Every test must trace to a result. One broken link produces a traceability gap, which is one of the top three reasons for FDA Major Deficiency letters. Spreadsheet-based traceability breaks under change. Purpose-built ALM tools (Jama Connect, Polarion, or Jira with compliance configuration) maintain trace links automatically.
3. Cybersecurity Across Connected Devices
Networked devices face active exploitation in healthcare environments. FDA now treats cybersecurity documentation as equivalent to safety documentation: a submission missing an SBOM or threat model is returned without substantive review.
4. Integrating AI and Machine Learning Responsibly
Data Governance: ML development models trained on undocumented datasets produce unreliable outputs across patient subgroups. FDA requires training data provenance and performance results stratified by demographic subgroup in AI/ML SaMD submissions.
Bias and Explainability: FDA reviewers expect evidence that AI outputs are interpretable in the clinical context. Black-box models face higher scrutiny in Class II and III submissions and require additional real-world performance monitoring commitments.
Model Monitoring: FDA's 2025 PCCP guidance allows approved AI components to be updated within predefined bounds without a new 510(k), but only when the change control plan was filed in the original submission. Teams that skip PCCP planning at the architecture phase lose this option.
5. Balancing Agile Development with Compliance Requirements
IEC 62304 does not require a waterfall. It requires documented outputs at each phase. Agile-compatible regulated development maps sprint artifacts to IEC 62304 deliverables: design inputs from the backlog, design reviews from sprint reviews, and test results from CI/CD pipelines. Teams that struggle are usually running Agile without phase output documentation.
6. Integrating Legacy Healthcare Systems
Many hospital environments run HL7 v2 interfaces, DICOM 3.0 stacks, and proprietary EHR APIs. New connected devices must interoperate with these systems. Also, they need to maintain audit trails and access controls that satisfy HIPAA and IEC 62304 SOUP requirements.
Best Practices for Building Regulatory-Compliant Medical Device Software
Here are a few patterns that correlate with first-submission approval and predictable market entry timelines:
|
Best Practice |
Concrete Action |
Outcome |
|
1. Classify before you code |
Determine the IEC 62304 class before architecture begins |
Prevents over- or under-documentation that adds months of rework |
|
2. Integrate risk management from Sprint 0 |
Add HARA items per feature; link to design controls and test cases |
Produces a live risk file that is audit-ready at submission |
|
3. Automate static analysis in CI/CD |
Polyspace/Coverity for embedded C; SAST for application layer |
FDA expects evidence of static analysis in Class B/C test reports |
|
4. Treat cybersecurity as a design input |
SBOM generation and STRIDE threat modeling before architecture sign-off |
Retrofitting costs 4 to 6x more; missing it returns submissions |
|
5. Build end-to-end traceability |
ALM tool linking requirements to design, to test, to result |
Closes the gap behind most FDA Major Deficiency letters |
|
6. Define post-market change control at v1.0 |
Document change process before the first release |
Enables emergency patches without process improvisation |
|
7. Plan AI governance and PCCP early |
Document model performance thresholds and change bounds in the original submission |
Reduces post-market regulatory burden for AI-enabled devices |
How to Select the Right Medical Device Software Development Partner?
The most expensive development partner is one that learns your regulatory framework on your timeline and budget. Evaluate these criteria before engagement.
|
Evaluation Area |
What to Verify? |
Why It Matters? |
|
Regulatory expertise |
IEC 62304 practitioner experience beyond awareness; ask for sample SDLC artifacts |
Reduces submission preparation time and deficiency risk |
|
Healthcare domain experience |
Prior FDA or MDR submissions; domain-specific architecture patterns |
Avoids architecture rework that compliance-naive teams generate |
|
Embedded, cloud, and AI capability |
Full-stack MedTech: firmware through cloud through ML pipeline |
Removes handoff risk between siloed vendors |
|
Validation process maturity |
Documented V&V process; ALM, static analysis, and test management tools in use |
Determines whether test output is submission quality from day one |
|
Cybersecurity competency |
SBOM generation, threat modeling, secure SDLC practices |
Non-negotiable for any networked device submission post-2023 |
|
Post-market support model |
Change management process, IEC 62304 section 6 compliance, incident response SLA |
Prevents emergency patch paralysis after product launch |
How does Your Team in India accelerate Medical Device Software Development?
Medical device software outsourcing used to mean compliance risk. Timezone gaps, regulatory inexperience, and engineers who knew code but not IEC 62304 were legitimate concerns a decade ago. But that profile no longer fits when you work with experts at Your Team in India. We bring you:
Advanced Healthcare and MedTech Engineering Capabilities
-
Embedded Software Development: Firmware development on ARM Cortex-M/A platforms, FreeRTOS and Embedded Linux, MISRA C compliance, and hardware-in-the-loop testing for Class B and C devices.
-
Cloud and Connected Device Platforms: AWS GovCloud and Azure Healthcare API deployments, HL7 FHIR R4 integration, DICOM imaging pipeline architecture, and encrypted device-to-cloud data flows with audit logging.
-
AI-Powered Healthcare Solutions: ML model development for diagnostic imaging, clinical decision support, and patient monitoring, with FDA PCCP documentation and post-market model monitoring built into the engagement scope.
-
Mobile Healthcare Applications: HIPAA-compliant iOS and Android applications with EHR/EMR integration via SMART on FHIR.
A Compliance-Focused Development Approach
-
IEC 62304-Aligned Workflows: Phase outputs are produced to submission standard from sprint one. Every artifact (SRS, architecture document, test report) is generated within an ISO 13485-aligned quality management system.
-
Documentation Support: Regulatory specialists co-author DHF deliverables alongside engineers. Clients don't arrive at submission with documentation gaps.
-
Validation and Verification Processes: Static analysis, automated test frameworks, and V&V protocols are configured per device class from the start of development.
Conclusion
Medical device software development is demanding because the standard for failure is measured in patient outcomes. Teams that reach market on schedule are not the ones that got lucky with regulators. They made compliance a design input, built cybersecurity and risk management into their architecture, and validated continuously rather than at submission.
AI-enabled devices add governance requirements, but they also represent the largest commercial opportunity in medical technology since connected devices became mainstream. FDA's PCCP framework creates a post-market update pathway for AI components, but only for teams that plan for it in the original submission.
The architecture and compliance decisions made in the first 60 days of development set the cost and timeline for everything that follows.

By Mangesh Gothankar

By Ashwani Sharma
By Achin Verma
Frequently Asked Questions
It is the process of designing, verifying, and maintaining software embedded in a medical device or operating independently for a medical purpose, under IEC 62304 and FDA or EU MDR.
IEC 62304 defines software lifecycle requirements for medical devices, classifying software by injury risk. FDA references it in all software guidance and expects 510(k) submissions to demonstrate conformance.
Multi-framework regulatory complexity, traceability gaps, mandatory cybersecurity documentation, AI/ML validation under probabilistic assumptions, and maintaining compliance within Agile sprint workflows cause most submission failures.
Embedded software (SiMD) runs on hardware inside a device, including pump controllers and pacemaker firmware. SaMD runs on general hardware for a medical purpose and follows a separate FDA pathway.
AI requires FDA PCCP documentation, training data provenance, and performance results by patient subgroup. Teams that include PCCP in the original submission can update models post-approval without a new 510(k).
A Class B SaMD built by a US team typically runs $800K to $2M through submission. A regulatory-experienced India-based team delivers equivalent scope at $350K to $900K.
Class A SaMD takes 6 to 9 months. Class B platforms take 12 to 18 months. Class C systems with AI components take 18 to 36 months.
Evaluate IEC 62304 practitioner experience, prior FDA or MDR submission history, full-stack firmware-to-cloud capability, documented V&V processes with ALM tooling, and a post-market change management model.






